
Il est urgent de mener des recherches novatrices pour prévenir l’apparition dévastatrice des maladies mitochondriales et améliorer la vie de ceux qui en sont déjà atteints.
Nous devons rassembler les esprits les plus brillants à travers le monde afin de développer de meilleures solutions pour soutenir la communauté Mito. L’excellence de la recherche en matière de santé est une force reconnue au Canada. Nous sommes connus dans le monde entier pour la rigueur et la réussite de nos essais cliniques. MitoCanada soutient des scientifiques incroyables. Nous vous invitons à faire leur connaissance et à découvrir leur travail.

Profil de découverte : Jessika Royea, PhD
MitoCanada s'associe à MITO2i pour financer la recherche sur les mitochondries qui pourrait déboucher sur de nouvelles thérapies pour [...]

Profil de découverte : Dr. Aneal Khan
MitoCanada finance la recherche pour faire progresser le diagnostic rapide de la maladie mitochondriale ( ) : Réduire les [...]

Profil de découverte : Dr. Mark Tarnopolsky
MitoCanada finance la recherche pour soutenir le développement de nouvelles thérapies pour traiter les maladies mitochondriales. Exerkines™sont [...]

Profil de découverte : Robert Screaton, PhD
MitoCanada s'associe à MITO2i pour financer la recherche sur les mitochondries qui pourrait déboucher surde nouvelles thérapies pour le [...]

Profil de découverte : Laura Rosella, PhD, MHSc
MitoCanada s'associe à MITO2i pour financer une bourse d'innovation visant à expliquer l'impact sur le système de santé de [...]

Profil de découverte : Dr. Ingrid Tein
MitoCanada s'associe à MITO2i pour financer la recherche sur les mitochondries qui pourrait améliorer la qualité de vie des [...]
MitoCanada soutient depuis longtemps la recherche dans sa quête de solutions pour les personnes qui vivent aujourd’hui avec une maladie mitochondriale. Certains des programmes de recherche que nous avons contribué à financer sont décrits ci-dessous.
Subventions accordées par le passé

Description de la bourse : Projet de recherche sur l’anémie sidéroblastique avec déficit immunitaire des cellules B, fièvres périodiques et retard de développement (SIFD) Montant de la bourse : 53 000 $ sur deux (2) ans. Investigateurs : Martin Holcik, Urszula Liwak-Muir ; Turaya Naas ; Quinlan Wylie, Hanna Ludwig , Ribal Kattini, Chloe Campbell, Brianna Rino, Romanah Ahmed, Megan Williams, et Angelo Slade*.
Étude de recherche :
Projet de recherche sur l’anémie sidéroblastique avec déficit immunitaire des cellules B, fièvres périodiques et retard de développement (SIFD). Ce projet de recherche visait à comprendre le mécanisme moléculaire qui explique pourquoi les mutations d’un gène appelé TRNT1 sont à l’origine de cette maladie mitochondriale rare mais dévastatrice. TRNT1 est une enzyme essentielle qui modifie tous les ARNt dans une cellule ; les ARNt matures sont, à leur tour, nécessaires à la synthèse correcte de toutes les protéines cellulaires. L’étude s’est concentrée sur un aspect particulier de la pathologie SIFD, à savoir le fait que les cellules dérivées des patients sont sensibles au stress oxydatif. L’hypothèse de travail était que la déficience en TRNT1 entraîne une réduction de la capacité à réparer les terminaisons des ARNt et donc une sensibilité accrue au stress oxydatif (et peut-être aussi à d’autres stress), contribuant ainsi au large phénotype observé chez les patients atteints de SIFD.
L’étude a identifié plusieurs protéines en aval qui sont exprimées de manière différentielle dans les cellules des patients. Les chercheurs ont également noté que les mêmes protéines sont exprimées de manière différentielle dans les tissus dérivés d’un modèle murin de TRNT1.
Cette étude de recherche a permis de former dix (10) stagiaires, deux (2) manuscrits ont été publiés et deux (2) sont en cours de préparation.
*Stagiaires : Urszula Liwak-Muir (boursière postdoctorale ; CHEO RI) ; Turaya Naas (boursière postdoctorale ; CHEO RI) ; Quinlan Wylie (étudiant de premier cycle, CHEO RI), Hanna Ludwig (étudiant de premier cycle, CHEO RI), Ribal Kattini (étudiant de premier cycle, Carleton), Chloe Campbell (étudiant de premier cycle, Carleton), Brianna Rino (étudiant de premier cycle, Carleton), Romanah Ahmed (étudiant de premier cycle, Carleton), Megan Williams (étudiante de premier cycle, Carleton), et Angelo Slade (étudiant en doctorat, Carleton).D., Carleton) Détails du financement : $53,686.81
par an pendant 2 ans.
Prix : Administration de cellules souches mésenchymateuses humaines comme traitement de la dysfonction mitochondriale. Montant de la subvention : 195 000 $ sur trois (3) ans Investigateurs : Chris Newell, Jane Shearer, Ph.D., Aneal Khan MSc, MD, FRCPC, FCCMG Étude de recherche : Les mitochondries sont les centrales énergétiques de la cellule, capables de générer une source continue d’ATP pour soutenir les fonctions métaboliques de base. Les maladies mitochondriales sont des troubles génétiques pathogènes qui touchent environ 1/3500-5000 individus, moins de 50% des enfants survivant jusqu’à l’âge adulte. Il n’existe actuellement aucun traitement ou remède efficace pour les maladies mitochondriales. Les cellules souches mésenchymateuses (CSM) sont des cellules multipotentes capables de se différencier en divers types de cellules. Leur capacité à cibler, moduler, régénérer et réparer les tissus a conduit à leur utilisation dans de multiples états pathologiques en pratique clinique. Ainsi, la recherche sur les cellules souches dans le domaine de la régénération des tissus endommagés a explosé, les études expérimentales et cliniques faisant état d’un potentiel thérapeutique passionnant. Des travaux récents de notre laboratoire et d’autres ont démontré que les CSM agissent en partie par fusion partielle, en transférant des mitochondries intactes des cellules vers le tissu hôte lésé, en modifiant l’expression des gènes mitochondriaux de l’hôte, en améliorant la capacité métabolique et en réduisant la production aberrante d’espèces réactives de l’oxygène (ROS). Tous ces mécanismes sont susceptibles d’améliorer la fonction métabolique de l’hôte, y compris la production d’ATP. Cependant, le taux de greffe de cellules dans les tissus de l’hôte est extrêmement faible, moins de 5 % des cellules survivant 24 heures après la transplantation. Notre laboratoire a montré que la manipulation du régime alimentaire (alimentation riche en graisses) favorise la greffe de cellules MSC dans divers tissus. Cette découverte a un potentiel énorme pour approfondir notre compréhension des facteurs qui favorisent le homing des cellules MSC et leur impact sur le métabolisme. La manipulation du régime alimentaire est également un traitement de première ligne pour certaines maladies mitochondriales.
DES OBJECTIFS SPÉCIFIQUES. L’objectif principal de ma thèse est d’utiliser les CSM comme outil pour influencer le métabolisme et traiter les dysfonctionnements mitochondriaux. Les données recueillies dans le cadre de cette thèse permettront d’examiner l’efficacité, la fonctionnalité et la longévité des mitochondries transférées des CSM à l’hôte, dans le cadre de la santé et de la maladie. Les objectifs spécifiques sont les suivants :
-
-
- Déterminer les mécanismes spécifiques par lesquels les CSM modifient la bioénergétique cellulaire et la fonction mitochondriale.
- Utiliser la manipulation du régime alimentaire pour favoriser la greffe de cellules dans les tissus de l’hôte.
- Examiner le potentiel thérapeutique des CSM dans un modèle de maladie mitochondriale.
-
HYPOTHÈSE GÉNÉRALE. La thérapie par cellules souches constitue une nouvelle modalité de traitement des maladies mitochondriales. L’hypothèse générale est que la thérapie par les CSM améliore la phosphorylation oxydative mitochondriale (OXPHOS), l’expression génétique et la capacité de piégeage des espèces réactives de l’oxygène (ROS) dans le foie. Ce résultat est obtenu en augmentant la densité des mitochondries fonctionnelles chez les animaux traités. Par conséquent, les animaux traités avec des CSM possèdent une fonction mitochondriale accrue, ce qui prouve le transfert de mitochondries à partir des CSM. Les niveaux de greffe cellulaire et de transfert mitochondrial seront plus élevés chez les animaux présentant un dysfonctionnement mitochondrial ainsi que chez ceux qui subissent un stress métabolique induit par un régime cétogène. Détails du financement : 105 000 $ (bourse de doctorat) avec des fonds de contrepartie de 30 000 $/an d’Alberta Innovates pour un total de 195 000 $ sur trois (3) ans.
Description du prix : Activation de la protéine kinase activée par l’AMP (AMPK) pour le traitement des maladies mitochondriales. Montant de la subvention : 210 000 $ sur deux (2) ans. Chercheurs : Alexander Green, M.Sc. (candidat au doctorat) ; Mark Tarnopolsky, M.D., Ph.D., FRCP(C) (superviseur), Neuromuscular and Neurometabolic Clinic, McMaster University Medical Center et Dr Gregory Steinberg, Ph.D. Étude de recherche :
La protéine kinase activée par l’AMP (AMPK) est une importante enzyme de détection de l’énergie. Normalement, l’AMPK est activée de manière endogène dans des conditions de stress énergétique par des kinases en amont et la liaison directe de l’AMP. Lorsqu’elle est active, l’AMPK active les voies permettant de limiter l’utilisation de l’énergie et d’augmenter la capacité de production d’énergie de la cellule. L’AMPK peut faciliter ce processus en augmentant le contenu mitochondrial et en éliminant les mitochondries dysfonctionnelles. De nouvelles thérapies pourraient être utilisées pour corriger le dysfonctionnement mitochondrial dans les maladies mitochondriales. Détails du financement :
35 000 $ par an pendant 2 ans avec des fonds de contrepartie du Programme de petits partenariats pour la santé (PPPOS) des Instituts de recherche en santé du Canada (IRSC) pour une bourse totale de 210 000 $ sur les 3 ans.
Description du prix: Utilisation d’un essai nouveau et unique pour cribler des bibliothèques de petits composés à la recherche de médicaments candidats pour améliorer la fonction mitochondriale en améliorant la fonction de la chaîne respiratoire dépendante de l’ubiquinone. Montant de la bourse : 45 000 $ sur un (1) an. Chercheurs : Siegfried Hekimi, Ph.D, Département de biologie, Université McGill, Montréal, Canada Étude de recherche :
Les maladies mitochondriales sont souvent des conditions dévastatrices dans lesquelles la fonction de production d’énergie des mitochondries est altérée par des mutations. L’un des types de défaut qui entraîne une telle altération est l’incapacité de l’organisme à synthétiser suffisamment d’ubiquinone (également appelée Co-enzyme Q, ou CoQ10). En outre, probablement parce que l’ubiquinone est synthétisée dans les mitochondries, les mitochondries malades deviennent moins capables de synthétiser suffisamment d’ubiquinone, ce qui aggrave leur état. Malheureusement, le simple fait de fournir de l’ubiquinone aux patients n’a que des effets minimes sur leur santé, probablement parce que l’ubiquinone exogène est mal absorbée par la plupart des organes du corps. Nous avons étudié la biosynthèse de l’ubiquinone dans un certain nombre d’organismes, y compris les souris. En particulier, nous avons développé des lignées de cellules cultivées incapables de produire de l’ubiquinone mais capables de se développer tant qu’on leur fournit du glucose, qu’elles peuvent utiliser pour produire suffisamment d’énergie sans dépendre de la fonction mitochondriale, qui est défectueuse en raison de l’absence d’ubiquinone. Ces cellules meurent rapidement en l’absence de glucose, mais peuvent être sauvées si on leur fournit de l’ubiquinone. Grâce à la subvention reçue de la Fondation MitoCanada, nous avons acheté et utilisé des bibliothèques de composés pour rechercher des médicaments susceptibles de sauver ou d’atténuer les défauts de production d’énergie dans les cellules déficientes en ubiquinone. Ces médicaments pourraient agir en stimulant la synthèse d’ubiquinone résiduelle, en remplaçant partiellement l’ubiquinone dans sa fonction dans les mitochondries, en augmentant l’activité ou l’efficacité de l’ubiquinone endogène résiduelle, ou même en stimulant la production d’énergie mitochondriale en l’absence d’ubiquinone. Notre criblage a déjà permis d’identifier un certain nombre de composés qui empêchent la mort des cellules déficientes en ubiquinone. Nous menons actuellement des expériences pour comprendre comment ces médicaments agissent pour produire leurs effets bénéfiques. Nous avons déjà développé une souche de souris qui perd progressivement l’ubiquinone de ses mitochondries et devient de plus en plus malade en conséquence. À long terme, nous pourrons tester nos médicaments candidats les plus prometteurs sur ces souris et déterminer ainsi dans quelle mesure ils pourraient aider les patients.
Prix : Calgary Next Generation DNA Sequencing Method for Mitochondrial Disease Diagnosis (Méthode de séquençage de l’ADN de nouvelle génération pour le diagnostic de la maladie mitochondriale ) Montant de la subvention : 100 000 $ sur trois (3) ans Investigateurs : Aneal Khan, MSc, MD, FRCPC, FCCMG Étude de recherche : Maladie mitochondriale chez les enfants à l’aide de la méthode de séquençage de nouvelle génération (NGS). La maladie mitochondriale entraîne un fonctionnement anormal du cœur, du cerveau, du foie et des muscles, ce qui se traduit par un développement insuffisant, des accidents vasculaires cérébraux, de multiples dysfonctionnements corporels et une durée de vie réduite. La maladie mitochondriale touche 1 personne sur 5000. Plus de 1 000 gènes sont impliqués dans la fonction mitochondriale et les méthodes de diagnostic précédentes impliquaient une biopsie invasive des tissus, ne pouvaient tester qu’une poignée de gènes, exposaient les patients à des risques de complications procédurales et ne pouvaient établir un diagnostic que dans un petit nombre de cas. Cependant, seuls 20 à 25 % des cas sont diagnostiqués à l’aide des méthodes traditionnelles de biopsie tissulaire, d’analyse biochimique et de séquençage des gènes candidats. Le projet NGS est une plateforme clinique utilisant une technologie de pointe pour diagnostiquer la maladie mitochondriale et est le premier du genre en Alberta et au Canada. La NGS ne nécessite pas de biopsies tissulaires douloureuses et risquées et peut être réalisée sur de l’ADN obtenu à partir d’un test sanguin ou d’un prélèvement de joue. L’originalité du projet de l’Alberta Children’s Hospital est d’apporter cette technologie au chevet des patients et de la rendre facile à utiliser par les médecins qui ont des patients atteints de maladies pour lesquelles le diagnostic est difficile à établir. En posant un diagnostic de maladie mitochondriale, les médecins peuvent prendre des décisions plus précises en matière de traitement, plutôt que d’attendre des mois ou des années pour obtenir une réponse à l’aide des méthodes traditionnelles. Grâce à un financement partiel de MitoCanada, le groupe a entrepris d’analyser les exomes de 30 patients albertains atteints de maladies mitochondriales non résolues. Lisez les données présentées sur 12 cas pilotes et le protocole recommandé pour 30 autres cas non diagnostiqués.
L’impact de la NGS sur la vie des patients atteints de maladie mitochondriale ne peut être surestimé. À titre d’exemple, grâce à la subvention accordée en 2012 par la CIBC, le Dr Khan a récemment identifié un gène responsable de la maladie mitochondriale chez une fillette de 8 ans qui a attendu toute sa vie qu’un diagnostic soit posé pour une maladie du foie dont elle a failli mourir à l’âge de 4 mois. On ne savait pas si les complications entraîneraient d’autres problèmes médicaux à l’avenir, ni ce que l’avenir lui réservait. Les données de la NGS ont permis d’identifier un gène qui s’est révélé être à l’origine des mêmes problèmes hépatiques dans la petite enfance, mais l’état de la plupart des enfants s’est progressivement amélioré avec le temps, ce qui signifie que les perspectives de rétablissement à long terme de cette enfant sont très bonnes et qu’elle n’a pas besoin de subir une autre biopsie du foie. Chez d’autres patients, dont certains étaient considérés comme atteints de sclérose en plaques, on a découvert une mutation génétique provoquant une maladie mitochondriale. Dans une autre famille, dont un enfant est décédé d’une maladie grave non diagnostiquée et dont le deuxième enfant présente la même détérioration, un gène semble avoir été identifié qui pourrait permettre un diagnostic et une méthode plus facile pour diagnostiquer la maladie chez d’autres enfants ou lors de futures grossesses. Le Dr Khan a reconnu la nécessité d’une approche différente pour le diagnostic précoce des maladies génétiques et métaboliques rares à Calgary. Poussé par le désir de voir un processus plus innovant et plus rapide pour le dépistage des patients, le Dr Khan a vu l’opportunité d’ouvrir un centre de recherche privé axé sur l’avancement des diagnostics des maladies génétiques rares et des maladies métaboliques héréditaires. En janvier 2016, il a ouvert la clinique Metabolics And Genetics In Calgary (M.A.G.I.C.), qui fait office de bureau de recherche hors site du Dr Khan, où son équipe effectue les procédures d’étude et le recrutement à partir des protocoles de recherche de l’Université de Calgary. Le financement de MitoCanada a été déterminant pour la création de cette clinique, qui offre des tests d’ADN gratuits (Mito-FIND) aux patients présentant des signes et/ou des symptômes de maladie mitochondriale déterminés par leur médecin. Plus d’informations ici : magiccalgary.ca Détails du financement : Financement de 100 000 $ sur trois ans. L’Alberta Children’s Hospital Foundation a fourni 50 000 dollars de fonds de contrepartie.
Description du prix: Séquençage de l’exome et de l’ARN comme outil de diagnostic moléculaire pour les maladies mitochondriales infantiles. Montant de la subvention : 45 000 $ sur deux (2) ans. Investigateurs : Eric A. Shoubridge(1), Isabelle Thiffault(2), Geneviève Bernard(3) Étude de recherche : Séquençage de l’exome et de l’ARN comme outil de diagnostic moléculaire pour les maladies mitochondriales infantiles ; intégration d’une stratégie informatique efficace et d’une analyse fonctionnelle pour caractériser la pathogénicité des variantes de l’ADN mitochondrial et des gènes nucléaires codant pour les protéines mitochondriales. Les tests de diagnostic moléculaire sont couramment utilisés pour confirmer les diagnostics cliniques des maladies mitochondriales héréditaires ; cependant, les diagnostics cliniques basés sur les séquences sont souvent indisponibles pour des gènes individuels ou des sous-groupes de maladies, et permettent rarement l’intégration des données de l’exome mitochondrial et de l’exome nucléaire. Le séquençage de nouvelle génération est un outil remarquable pour la découverte de nouveaux gènes pathologiques, mais il reste difficile d’interpréter les variations de l’ADN chez les patients. Un nombre croissant de gènes sont associés aux maladies mitochondriales infantiles, et comme plus de 1000 protéines participent à la biologie mitochondriale, nous nous attendons à ce que de nombreux autres gènes pathogènes restent à découvrir. Ce projet de recherche vise à produire des tests génétiques diagnostiques complets pour les troubles mitochondriaux, à évaluer l’impact des gènes modificateurs sur l’apparition et la gravité de la maladie, et à étudier la pathogenèse de différents troubles mitochondriaux par une analyse fonctionnelle des lignées cellulaires obtenues à partir de patients. Détails du financement : 45 000 $ pour deux ans. (Ce projet est également soutenu par l’hôpital Children’s Mercy de Kansas City).
Description du prix : Une plateforme intégrée de diagnostic mitochondrial combinant le séquençage de nouvelle génération avec des études respiratoires dans des cellules de patients Montant de la subvention : 87 000 $ sur un (1) an. Investigateurs : Matthew A. Lines MD MSc1, Dennis Bulman PhD1,2, Pranesh Chakraborty MD1,2, C. Anthony Rupar PhD3,4, Martin HolcikPhD2, Mary-Ellen Harper PhD5 Étude de recherche : Les maladies mitochondriales représentent un groupe diversifié d’affections génétiques qui se sont toujours révélées difficiles à diagnostiquer, à étudier et à traiter en raison de leur degré élevé d’hétérogénéité génétique. Bien que le séquençage de nouvelle génération (NGS) permette facilement le séquençage simultané des >1000 gènes mitochondriaux connus codés dans les génomes nucléaire et mitochondrial, l’interprétation des données est compliquée par (i) l’abondance des variants codants rares détectés chez un individu donné, et (ii) le manque de connaissances concernant les fonctions et les phénotypes de nombreux gènes mitochondriaux. Les premières expériences suggèrent que les tests basés sur la NGS permettront d’identifier un gène de maladie mitochondriale connue chez environ 25 % des patients probants bien sélectionnés ; environ 25 % supplémentaires présentent des changements dans de nouveaux gènes candidats nécessitant une évaluation fonctionnelle pour la validation de la pathogénicité. Afin d’améliorer l’accès des Canadiens atteints de maladies mitochondriales à cette technologie cruciale, nous avons constitué un groupe de travail translationnel composé d’experts en métabolisme clinique, en identification de gènes de maladies par NGS et en physiologie/bioénergétique mitochondriale. Nous proposons une étude pilote translationnelle de diagnostic mitochondrial par NGS, soutenue par un panel d’essais fonctionnels dans des cellules de patients pour évaluer la respiration, la morphologie et la dynamique mitochondriales, et l’état redox cellulaire. L’intégration d’informations fonctionnelles nous permettra (i) de corroborer, d’infirmer et/ou d’affiner les hypothèses cliniques suggérées par les données NGS, et (ii) d’évaluer l’efficacité in vitro de divers substrats, cofacteurs et médicaments sur des cellules intactes de patients. Les connaissances méthodologiques acquises au cours de l’étude contribueront à la mise en place d’une ressource diagnostique permanente, et les données (nouveaux gènes pathologiques) serviront de tremplin à plusieurs autres projets de science fondamentale. Les données concernant le profil de réponse in vitro aux médicaments et aux cofacteurs de chaque affection spécifique (gène) seront utilisées pour encadrer d’autres études de thérapie sur mesure. Détails du financement : Financement de 32 000 $ pour un an. Le Centre hospitalier pour enfants de l’est de l’Ontario a fourni un financement de contrepartie de 55 000 $.
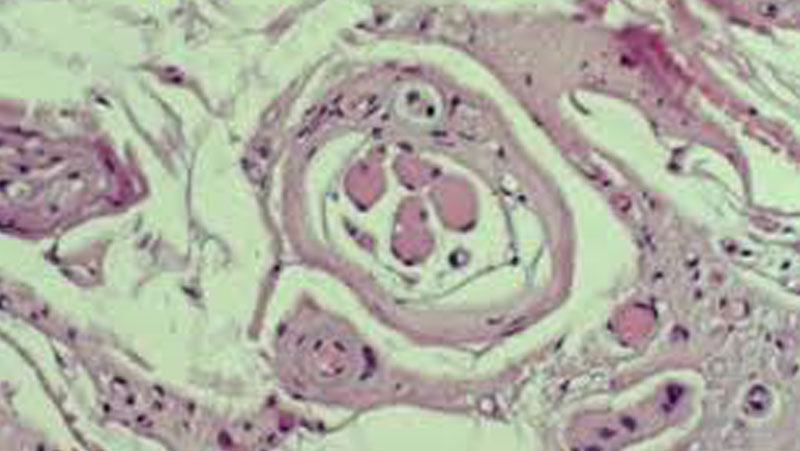 Prix : Congelé ou frais : Quel type d’échantillon musculaire est le plus efficace pour le diagnostic de la maladie mitochondriale dans un laboratoire clinique ? Montant de la subvention : 210 000 $ sur trois (3) ans. Chercheurs : Lauren MacNeil PhD 1, Murray Potter MD 1,3, Mark Tarnopolsky, MD, PhD2,4 Étude de recherche : Le diagnostic de la maladie mitochondriale est établi à l’aide de tests biochimiques effectués sur une biopsie musculaire. La mesure de la fonction mitochondriale est plus précise lorsqu’elle est effectuée sur des échantillons frais peu de temps après le prélèvement. Cependant, de nombreux échantillons sont prélevés dans des endroits éloignés d’un centre de diagnostic et doivent être congelés pour être stockés/expédiés. La congélation limite les types d’analyses possibles et peut introduire des facteurs susceptibles d’altérer les résultats des tests et la capacité à détecter correctement un dysfonctionnement mitochondrial. À l’aide d’échantillons musculaires prélevés sur des participants adultes atteints ou non d’une maladie mitochondriale, nous comparerons la respirométrie haute résolution et l’activité enzymatique dans le tissu frais à l’activité protéique et enzymatique dans le tissu congelé afin de déterminer si l’une ou l’autre de ces méthodes est supérieure. Le diagnostic d’une maladie mitochondriale est réalisé à l’aide de tests biochimiques effectués sur une biopsie musculaire. La mesure de la fonction mitochondriale est plus précise lorsqu’elle est effectuée sur des échantillons frais peu de temps après le prélèvement. Cependant, de nombreux échantillons sont prélevés dans des endroits éloignés d’un centre de diagnostic et doivent être congelés pour être stockés/expédiés. La congélation limite les types d’analyses possibles et peut introduire des facteurs susceptibles d’altérer les résultats des tests et la capacité à détecter correctement un dysfonctionnement mitochondrial. À l’aide d’échantillons musculaires prélevés sur des participants adultes atteints ou non d’une maladie mitochondriale, nous comparerons la respirométrie haute résolution et l’activité enzymatique dans le tissu frais à la protéine et à l’activité enzymatique dans le tissu congelé afin de déterminer si l’une ou l’autre méthode est supérieure. Tout d’abord, les chercheurs souhaitent remercier MitoCanada pour le financement généreux de ce projet de recherche et lui faire savoir qu’il y aura un bénéfice direct à long terme qui s’étendra au-delà de la recherche. C’est en grande partie grâce à ce projet que le Dr Lauren MacNeil s’est vu attribuer cette année l’une des deux très convoitées bourses de formation en laboratoire de génétique clinique du ministère de la Santé et des Soins de longue durée de l’Ontario. Cette bourse financera sa formation dans le cadre du programme de formation en génétique biochimique du Collège canadien des généticiens médicaux à l’Université McMaster pendant les deux prochaines années. Depuis que nous avons reçu le financement de l’étude susmentionnée, nous avons recruté 34 participants souffrant de troubles musculaires et 17 hommes en bonne santé. Les tissus musculaires frais ont été étudiés par respirométrie à haute résolution et par activité enzymatique. La collecte des échantillons vient de s’achever, les essais enzymatiques sur les tissus congelés et l’analyse des données sont encore en cours ; cependant, nous avons fait plusieurs observations préliminaires et nous présenterons ces résultats lors de réunions scientifiques consacrées aux erreurs innées du métabolisme et aux maladies mitochondriales. L’un de ces rapports, qui porte sur la stabilité des échantillons de muscle après leur prélèvement, sera présenté au symposium Garrod qui se tiendra en mai à Vancouver. Il est bien connu que, si elle n’est pas manipulée correctement, l’activité des complexes de la chaîne respiratoire diminue avec le temps, mais l’effet relatif de la manipulation des échantillons sur la respirométrie à haute résolution et les essais d’activité enzymatique n’est pas connu.
Prix : Congelé ou frais : Quel type d’échantillon musculaire est le plus efficace pour le diagnostic de la maladie mitochondriale dans un laboratoire clinique ? Montant de la subvention : 210 000 $ sur trois (3) ans. Chercheurs : Lauren MacNeil PhD 1, Murray Potter MD 1,3, Mark Tarnopolsky, MD, PhD2,4 Étude de recherche : Le diagnostic de la maladie mitochondriale est établi à l’aide de tests biochimiques effectués sur une biopsie musculaire. La mesure de la fonction mitochondriale est plus précise lorsqu’elle est effectuée sur des échantillons frais peu de temps après le prélèvement. Cependant, de nombreux échantillons sont prélevés dans des endroits éloignés d’un centre de diagnostic et doivent être congelés pour être stockés/expédiés. La congélation limite les types d’analyses possibles et peut introduire des facteurs susceptibles d’altérer les résultats des tests et la capacité à détecter correctement un dysfonctionnement mitochondrial. À l’aide d’échantillons musculaires prélevés sur des participants adultes atteints ou non d’une maladie mitochondriale, nous comparerons la respirométrie haute résolution et l’activité enzymatique dans le tissu frais à l’activité protéique et enzymatique dans le tissu congelé afin de déterminer si l’une ou l’autre de ces méthodes est supérieure. Le diagnostic d’une maladie mitochondriale est réalisé à l’aide de tests biochimiques effectués sur une biopsie musculaire. La mesure de la fonction mitochondriale est plus précise lorsqu’elle est effectuée sur des échantillons frais peu de temps après le prélèvement. Cependant, de nombreux échantillons sont prélevés dans des endroits éloignés d’un centre de diagnostic et doivent être congelés pour être stockés/expédiés. La congélation limite les types d’analyses possibles et peut introduire des facteurs susceptibles d’altérer les résultats des tests et la capacité à détecter correctement un dysfonctionnement mitochondrial. À l’aide d’échantillons musculaires prélevés sur des participants adultes atteints ou non d’une maladie mitochondriale, nous comparerons la respirométrie haute résolution et l’activité enzymatique dans le tissu frais à la protéine et à l’activité enzymatique dans le tissu congelé afin de déterminer si l’une ou l’autre méthode est supérieure. Tout d’abord, les chercheurs souhaitent remercier MitoCanada pour le financement généreux de ce projet de recherche et lui faire savoir qu’il y aura un bénéfice direct à long terme qui s’étendra au-delà de la recherche. C’est en grande partie grâce à ce projet que le Dr Lauren MacNeil s’est vu attribuer cette année l’une des deux très convoitées bourses de formation en laboratoire de génétique clinique du ministère de la Santé et des Soins de longue durée de l’Ontario. Cette bourse financera sa formation dans le cadre du programme de formation en génétique biochimique du Collège canadien des généticiens médicaux à l’Université McMaster pendant les deux prochaines années. Depuis que nous avons reçu le financement de l’étude susmentionnée, nous avons recruté 34 participants souffrant de troubles musculaires et 17 hommes en bonne santé. Les tissus musculaires frais ont été étudiés par respirométrie à haute résolution et par activité enzymatique. La collecte des échantillons vient de s’achever, les essais enzymatiques sur les tissus congelés et l’analyse des données sont encore en cours ; cependant, nous avons fait plusieurs observations préliminaires et nous présenterons ces résultats lors de réunions scientifiques consacrées aux erreurs innées du métabolisme et aux maladies mitochondriales. L’un de ces rapports, qui porte sur la stabilité des échantillons de muscle après leur prélèvement, sera présenté au symposium Garrod qui se tiendra en mai à Vancouver. Il est bien connu que, si elle n’est pas manipulée correctement, l’activité des complexes de la chaîne respiratoire diminue avec le temps, mais l’effet relatif de la manipulation des échantillons sur la respirométrie à haute résolution et les essais d’activité enzymatique n’est pas connu.
Notre premier rapport sur ce sujet présente nos observations sur la stabilité du tissu musculaire conservé jusqu’à une semaine après l’excision pour la respirométrie à haute résolution. L’activité du complexe I a été la première et la plus affectée par le temps, chutant de 45% en 24 heures et atteignant une perte de fonction presque complète à 7 jours. Les complexes II et III sont restés stables pendant 72 heures, mais au 7e jour, leurs activités étaient respectivement inférieures de 44 % et de 50 %. Le complexe IV est resté stable pendant 7 jours. Ces résultats indiquent qu’une mesure immédiate après l’excision serait impérative pour rendre compte de l’activité de l’IC par respirométrie à haute résolution. Une autre observation sera présentée en juin lors du symposium de la United Mitochondrial Disease Foundation à Washington. Plusieurs patients ayant participé à l’étude présentaient une myosite ou une polymyosite à corps inclus. Il s’agit de myopathies inflammatoires avec inflammation musculaire, faiblesse musculaire et dysfonctionnement mitochondrial. La respirométrie à haute résolution chez ces patients a montré un schéma spécifique : un flux très faible du complexe I et du complexe I + II, et une augmentation paradoxale du flux du complexe II à un niveau plus élevé que le flux du complexe I + II lorsque le complexe I est inhibé. Cela implique que le complexe I entrave l’activité maximale du complexe II chez ces patients et laisse entrevoir de nouveaux domaines à explorer pour comprendre et traiter ces troubles. Détails du financement : Financement de 210 000 $ pour trois ans avec un financement de contrepartie du Programme de partenariat pour la petite santé (PPPOS) des Instituts de recherche en santé du Canada (IRSC).
Prix : Stratégies visant à améliorer le transfert de mitochondries par les cellules souches : Potentiel pour le traitement de la maladie mitochondriale. Montant de la subvention : 45 000 $ sur un (1) an. Chercheurs : Jane Shearer, Ph.D. ; Jong Rho, MD ; Steven Martin, MD ; Aneal Khan, MD Étude de recherche : Cette étude examine l’utilité d’une thérapie cellulaire (cellules souches) pour atténuer les symptômes associés au dysfonctionnement mitochondrial. Les cellules souches sont prometteuses car elles sont capables de réduire le stress oxydatif, de soulager l’inflammation et de réparer les tissus endommagés. Des travaux récents montrent également qu’elles ont la capacité de transférer des mitochondries saines dans les tissus hôtes. L’objectif de cette recherche est de mieux comprendre ce processus et son utilité clinique potentielle pour le traitement des maladies mitochondriales. L’équipe de recherche est unique en son genre, car elle combine des compétences en matière de maladie mitochondriale, de physiologie métabolique, de neurologie pédiatrique et de transplantation de cellules. Avec le soutien de MitoCanada, nous espérons que nos travaux contribueront un jour à la mise au point d’une thérapie cellulaire pour le traitement des maladies mitochondriales. Les maladies mitochondriales sont des affections rares, mais souvent dévastatrices, dont les options thérapeutiques sont limitées. La nature variée et souvent multiforme des maladies mitochondriales exige une approche qui permette de délivrer ou de stimuler la prolifération de mitochondries saines. Une nouvelle approche pour atteindre cet objectif implique l’administration de cellules souches. Les cellules souches sont des cellules précoces à lignées multiples (qui peuvent se transformer en de nombreux types de cellules) et bénéficient d’un privilège immunitaire (elles ne sont pas attaquées par le système immunitaire). Tel était l’objectif de notre récent projet financé par MitoCanada, intitulé « Stratégies visant à améliorer le transfert de mitochondries par les cellules souches : potentiel pour le traitement des maladies mitochondriales ». Dans cette étude, nous montrons que les cellules souches ciblent les sites de dysfonctionnement mitochondrial et réduisent une caractéristique clé associée à la maladie, la production d’espèces réactives de l’oxygène en excès. Cette réduction s’est produite 5 jours après l’administration, malgré le fait que la majorité des cellules souches injectées n’étaient plus présentes dans le foie, le principal tissu d’intérêt. Ces travaux ont été publiés récemment. Pour plus d’informations, cliquez ici. Pour l’avenir, nous essayons de découvrir « comment » cela se produit. Alors que le laboratoire avait initialement émis l’hypothèse d’un transfert direct de mitochondries des cellules souches vers les tissus hôtes, nous montrons aujourd’hui que les cellules souches modifient l’expression des gènes et les réseaux mitochondriaux, ce qui entraîne un passage du métabolisme mitochondrial au métabolisme glycolytique (du grec « glyco » signifiant « doux » et du grec « lysein » signifiant « briser en morceaux »). Ce changement entraîne une diminution de la dépendance des mitochondries pour la production d’énergie et réorganise les mitochondries pour favoriser les mitochondries saines existantes. Pour ce faire, nous collaborons avec le Dr Dustin Hitteland du National Children’s Hospital de Washington, DC, afin d’examiner les profils d’expression génétique de nos échantillons. Cela permettra de comprendre comment les cellules souches induisent ce changement. Les données résultant de cette étude amélioreront notre compréhension des mécanismes de régénération à médiation cellulaire et le développement éventuel d’un nouveau traitement pour la maladie mitochondriale. Détails du financement : Financement de 45 000 $ pour un an.
Prix : Identification des facteurs sériques responsables de la biogenèse mitochondriale systémique Montant de la subvention : 41 000 $ sur un (1) an Investigateurs : Mark Tarnopolsky, M.D., Ph.D., FRCP(C) ; Justin Crane, M.Sc. ; Adeel Safdar, Ph.D. ; Arkan Abadi, Ph.D. Étude de recherche : Nous avons montré que l’exercice d’endurance à long terme offrait une protection clinique spectaculaire dans pratiquement tous les tissus du corps d’un modèle murin de maladie mitochondriale (souris POLG1). La protection s’étendait à des tissus qui ne sont pas traditionnellement associés aux bienfaits de l’exercice, notamment les gonades, la peau, la fourrure, les yeux, les poumons et les reins. Cela indique qu’un facteur systémique libéré en réponse à l’exercice agit sur des tissus éloignés, un peu comme une hormone. La recherche financée par MitoCanada nous permettra d’identifier plus précisément ces facteurs. Le financement soutiendra en partie deux études majeures. La première est une étude transversale portant sur 100 hommes et femmes âgés de 20 à 86 ans, dont la moitié sont des athlètes bien entraînés et l’autre moitié des témoins sédentaires. L’analyse de la fonction mitochondriale sera effectuée sur des cellules de la joue, de la peau, des sédiments urinaires (cellules rénales) et du sang afin de déterminer si les avantages systémiques que nous avons observés chez les souris s’appliquent également aux humains. Le deuxième aspect consistera à essayer d’identifier les facteurs qui confèrent une protection mitochondriale systémique. Pour ce faire, nous cultiverons des cellules de peau en présence de sérum provenant d’humains au repos et après l’exercice. Tout d’abord, nous identifierons le moment de la période post-exercice où le sérum a un effet bénéfique sur la biogenèse mitochondriale et nous utiliserons ce sérum pour l’analyse protéomique (identification des protéines) qui sont responsables de l’effet. L’objectif à long terme est d’identifier une ou plusieurs protéines qui pourraient être injectées aux patients qui ne peuvent pas faire d’exercice pour protéger les mitochondries dans plusieurs tissus. Dans l’ensemble, ces résultats devraient avoir des implications significatives pour le vieillissement humain, la physiologie musculaire, la science de l’exercice et, surtout, la médecine mitochondriale.