
The Mito Cocktail


Some mitochondrial specialists may recommend a combination of vitamins and mitochondrial cofactors (nutrients that help enzymes do their jobs), often referred to as the mito cocktail. The goal of a mito cocktail is supportive, to help optimize mitochondrial function, reduce metabolic stress, and improve symptom management.
It’s important to know that there is no single standard mito cocktail. Supplement choices (if recommended at all) depend on an individual’s diagnosis, symptoms, age, medications and lab findings. Clinical experts emphasize that while strong randomized trial evidence is limited for many supplements, supplements are often used in practice because some have biologic rationale, relatively favourable safety profiles when monitored, and may be particularly relevant in specific mito conditions.
![]()
Specialists may consider supplements because they can help:
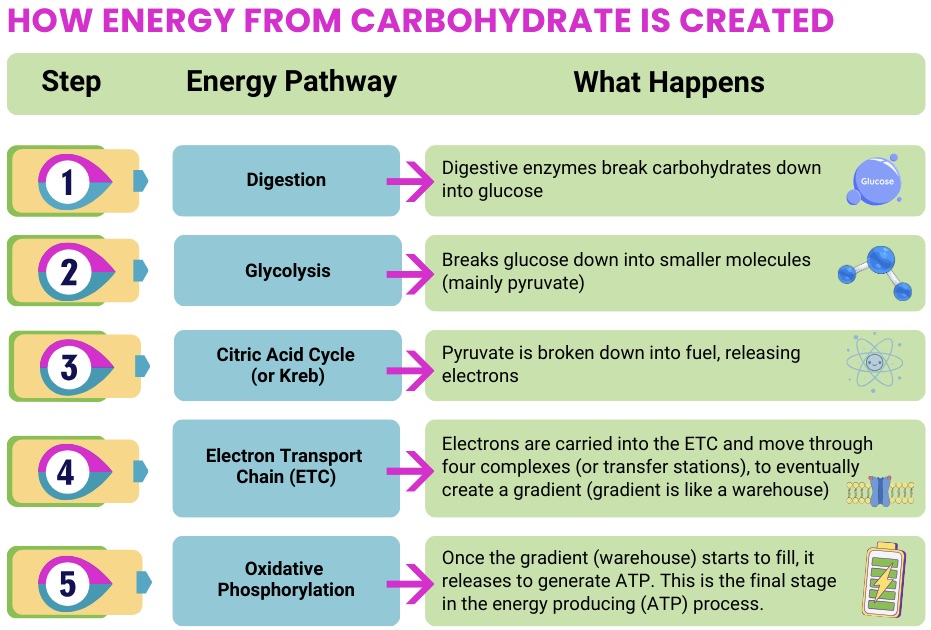
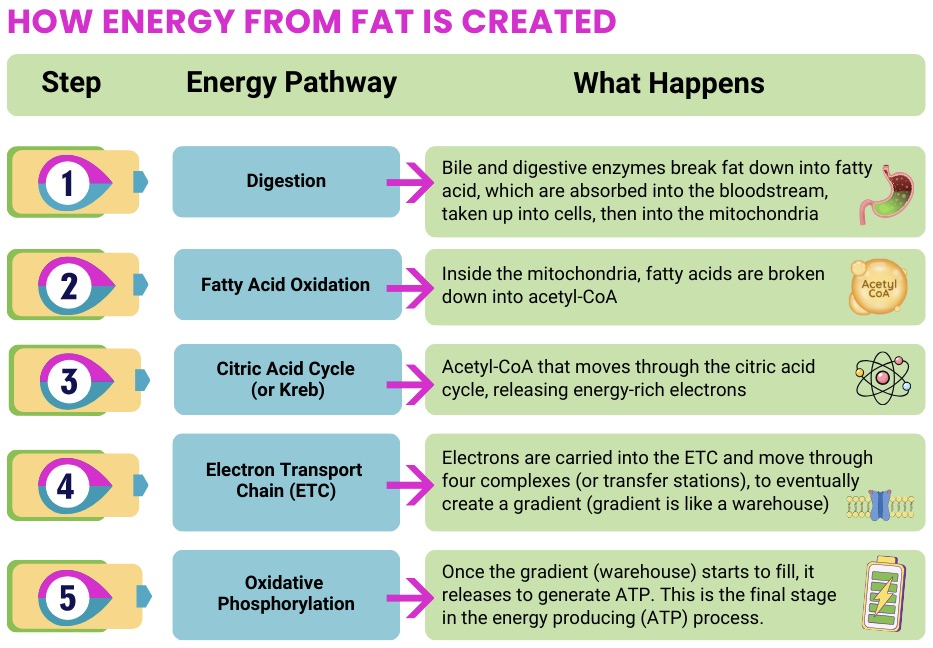
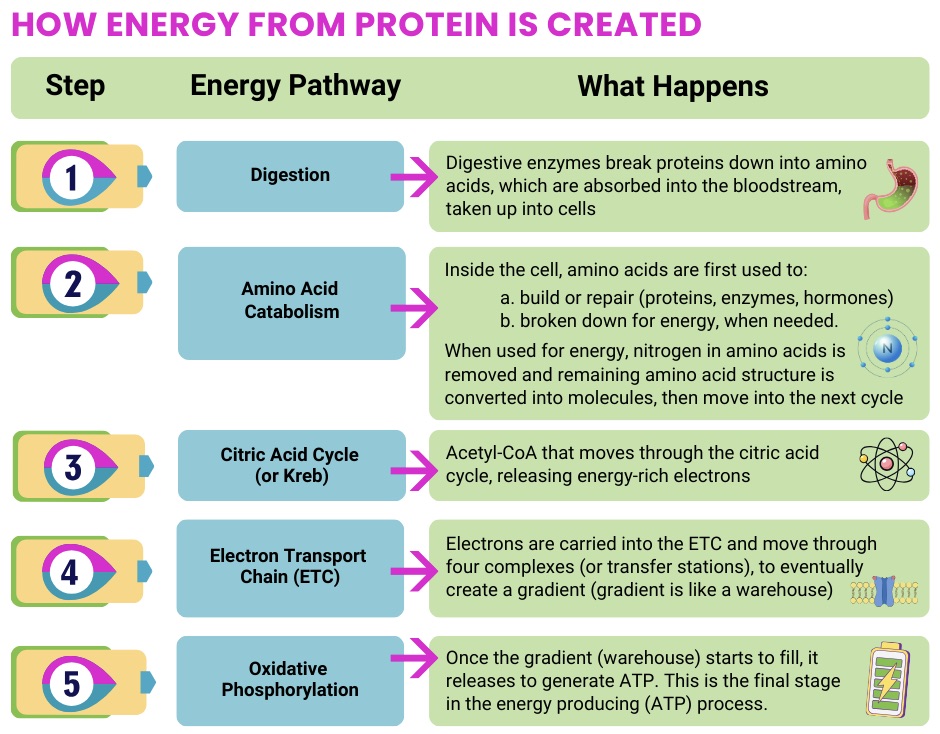
- Support energy production pathways (including oxidative phosphorylation)
- Provide cofactors needed for mitochondrial enzymes
- Help manage secondary effects of mitochondrial dysfunction (like oxidative stress)
- Address suspected or documented nutrient deficiencies
![]()
In some mitochondrial disorders, specific supplements are more commonly considered because of the underlying biology. For example, the Mitochondrial Medicine Society care standards note that riboflavin may be considered in ACAD9-related myopathy and that a combination of CoQ10 and riboflavin may be considered for ETFDH gene-related myopathy.
![]()
These are often discussed during mito clinc appointments and in patient resources, but not everyone needs them, and dosing/selection should be individualized.

![]()
There’s a fair amount of supplement information online that can sound very convincing. But with mitochondrial disease, the right supplement plan depends on the type of diagnosis and the right clinical context. Supplements can interact with medications, affect lab test results and, depending on the individual, may cause side effects.

Before starting, stopping or changing any supplement, it’s strongly recommended that you discuss it with the specialist overseeing your mitochondrial disease care.
















 Launching a New Era in Mitochondrial Gene Editing
Launching a New Era in Mitochondrial Gene Editing Sometimes happenstance brings about the most meaningful paths. “I started working on mitochondrial diseases by pure chance, but the more I worked on it, the more I fell in love with it,” says Dr. Moraes. “The mitochondrion is like a battery inside the cell, and it has its own DNA. It’s the only organelle besides the nucleus that does. It’s a fascinating system, and so I’ve devoted my career to it.”
Sometimes happenstance brings about the most meaningful paths. “I started working on mitochondrial diseases by pure chance, but the more I worked on it, the more I fell in love with it,” says Dr. Moraes. “The mitochondrion is like a battery inside the cell, and it has its own DNA. It’s the only organelle besides the nucleus that does. It’s a fascinating system, and so I’ve devoted my career to it.” Continuing his research, Dr. Moraes and his colleagues found that a specific genetic mutation, usually responsible for mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), also caused a variety of manifestations. “This mutation is one of the most common mtDNA mutations in the patient population,” he says. “In 1993, we published research showing that patients with this mutation could have many different types of diseases and many different symptoms, and that these symptoms clustered within families, suggesting that nuclear DNA plays a role in modifying how the mtDNA mutation shows up.”
Continuing his research, Dr. Moraes and his colleagues found that a specific genetic mutation, usually responsible for mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), also caused a variety of manifestations. “This mutation is one of the most common mtDNA mutations in the patient population,” he says. “In 1993, we published research showing that patients with this mutation could have many different types of diseases and many different symptoms, and that these symptoms clustered within families, suggesting that nuclear DNA plays a role in modifying how the mtDNA mutation shows up.”
 research, Dr. Moraes and his lab used one of the base editors to rescue mitochondrial function in a mouse model. “We found a way to base edit a gene with a pathogenic mutation so that it became stable, improving the function of the mitochondrial energy production in the mouse model,” he says.
research, Dr. Moraes and his lab used one of the base editors to rescue mitochondrial function in a mouse model. “We found a way to base edit a gene with a pathogenic mutation so that it became stable, improving the function of the mitochondrial energy production in the mouse model,” he says. a thick skin and to be tenacious. “We have to keep pushing,” he says. “There are lots of failures in this field, but a failure isn’t a total failure if you understand why the experiment didn’t work. It always teaches you something.”
a thick skin and to be tenacious. “We have to keep pushing,” he says. “There are lots of failures in this field, but a failure isn’t a total failure if you understand why the experiment didn’t work. It always teaches you something.”")

")
")
")
")
")
")