
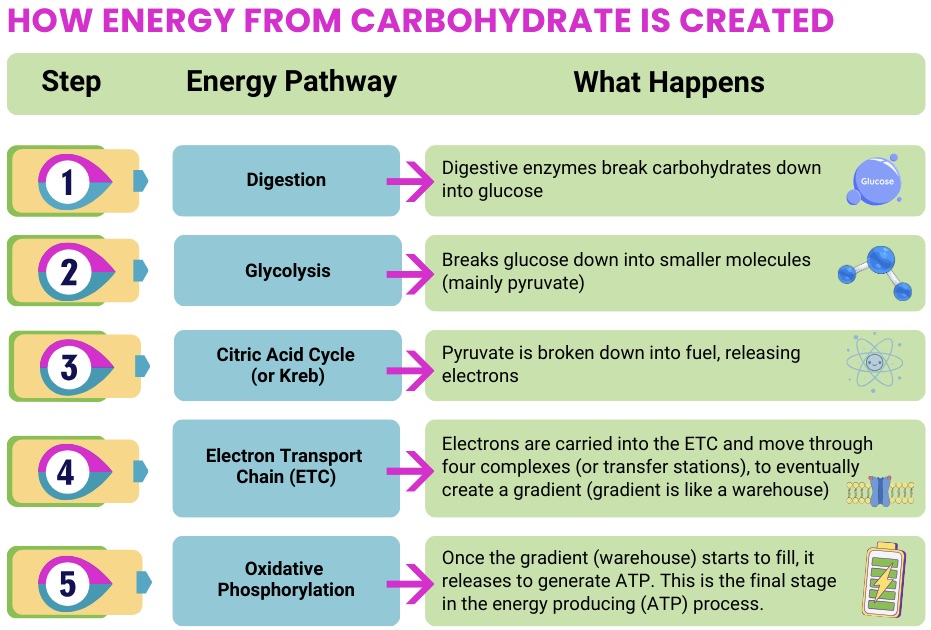
Nutrition and Specific Mito Disorders


As mitochondrial disease represents over 350 different diagnoses, it makes sense that nutrition can play different roles depending on the underlying condition.
In some disorders, nutrition strategies are focused on preventing metabolic crisis; in others, the goal may be to reduce catabolic stress, support muscle health, or complement diagnosis-specific therapies. Broad clinical care standards emphasize individualized planning and avoiding metabolic stressors (like prolonged fasting), with diagnosis-specific guidance helping further shape nutrition approaches.
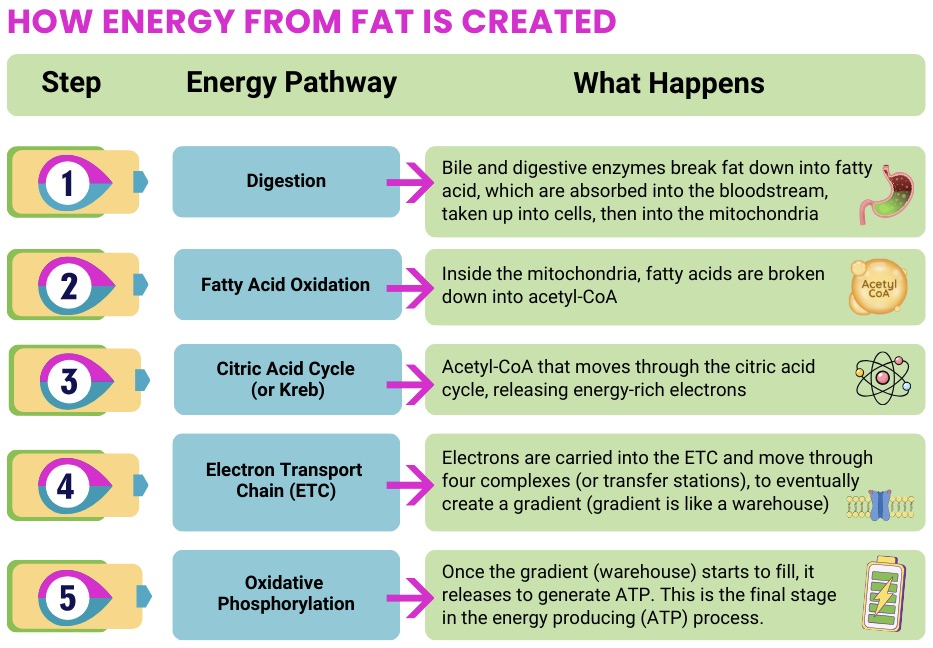
For FAOD, nutrition is often a central part of disease management. Because the body many not be able to use fats effectively for energy, maintaining a reliable fuel supply (often emphasizing carbohydrates) and avoiding fasting is critical. Clinical and guideline literature repeatedly highlights:

MELAS can involve high metabolic demand and multi-system symptoms (including GI and nutrition challenges), so nutrition strategies often emphasize maintaining energy balance and preventing catabolic stress during illness or poor intake.
Targeted supplementation is sometimes discussed in MELAS, most notably arginine and/or citrulline in relation to stroke-like episodes. The literature includes supportive reports and reviews, but consensus guidance also notes limitations and ongoing debate about strength of evidence and clinical use.
In MELAS, nutrition care often focuses on steady intake, illness-day planning, and discussing targeted therapies (like specific supplements) with a mitochondrial specialist because recommendations vary across clinics and individuals.
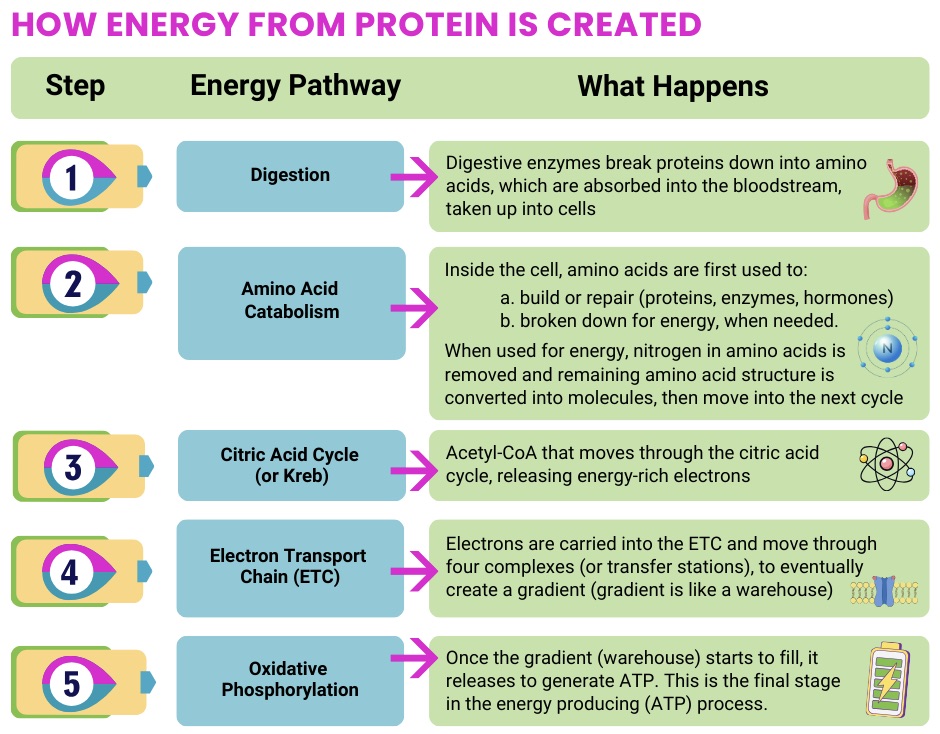
When muscle involvement is prominent, nutrition strategies often focus on supporting muscle maintenance and minimizing fatigue by ensuring adequate overall calories and protein, especially if appetite is low or unintentional weight loss is present.
Some research in mitochondrial disease populations (including those with muscle symptoms) suggests that insufficient intake is common, and that higher intakes of macronutrients (including protein) may correspond with better outcomes like muscle strength, lower fatigue, and improved quality of life, though this doesn’t mean “more is always better,” and personalization matters.
For mitochondrial myopathies, nutrition support often prioritizes “enough energy + enough protein,” using practical strategies (small frequent meals, energy-dense additions, smoothies) when fatigue or GI issues make intake difficult.
![]()
POLG-related disorders can present very differently from one person to another. Nutrition strategies commonly emphasize preventing catabolic stress (especially during illness or reduced intake) and maintaining consistent energy availability, an approach aligned with broader mitochondrial care standards.
In some POLG-related conditions, seizures can be a major concern, and dietary therapies (such as ketogenic-style approaches) may be considered in specialized contexts (typically for difficult-to-treat refractory epilepsy) under expert supervision, with careful monitoring and individualized risk–benefit discussions.
For POLG, the key nutrition themes are often “avoid catabolic stress, plan for illness, and create individualize dietary approaches with your mito specialist and care team,” especially if seizures or significant GI/nutrition issues are part of the picture.
![]()


Because mitochondrial disease includes many different conditions, nutrition approaches are rarely one-size-fits-all. Some disorders rely heavily on nutrition strategies to prevent metabolic crisis, while others focus on maintaining energy balance, supporting muscle health, or managing symptoms. Across many diagnoses, maintaining consistent energy intake and avoiding metabolic stress are key themes. Individualized guidance from a mitochondrial specialist or dietitian is important to determine the best approach.






















 Launching a New Era in Mitochondrial Gene Editing
Launching a New Era in Mitochondrial Gene Editing Sometimes happenstance brings about the most meaningful paths. “I started working on mitochondrial diseases by pure chance, but the more I worked on it, the more I fell in love with it,” says Dr. Moraes. “The mitochondrion is like a battery inside the cell, and it has its own DNA. It’s the only organelle besides the nucleus that does. It’s a fascinating system, and so I’ve devoted my career to it.”
Sometimes happenstance brings about the most meaningful paths. “I started working on mitochondrial diseases by pure chance, but the more I worked on it, the more I fell in love with it,” says Dr. Moraes. “The mitochondrion is like a battery inside the cell, and it has its own DNA. It’s the only organelle besides the nucleus that does. It’s a fascinating system, and so I’ve devoted my career to it.”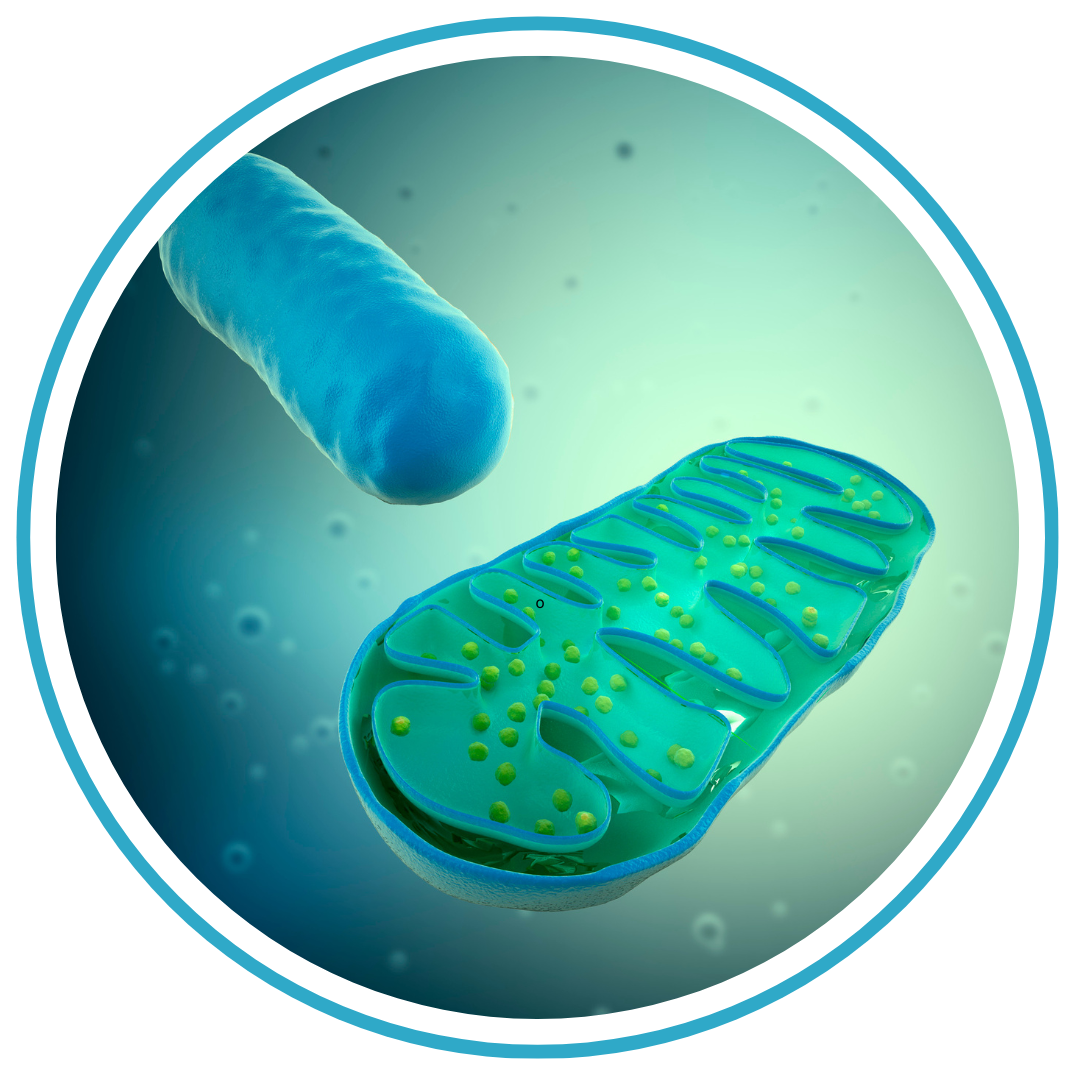 Continuing his research, Dr. Moraes and his colleagues found that a specific genetic mutation, usually responsible for mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), also caused a variety of manifestations. “This mutation is one of the most common mtDNA mutations in the patient population,” he says. “In 1993, we published research showing that patients with this mutation could have many different types of diseases and many different symptoms, and that these symptoms clustered within families, suggesting that nuclear DNA plays a role in modifying how the mtDNA mutation shows up.”
Continuing his research, Dr. Moraes and his colleagues found that a specific genetic mutation, usually responsible for mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), also caused a variety of manifestations. “This mutation is one of the most common mtDNA mutations in the patient population,” he says. “In 1993, we published research showing that patients with this mutation could have many different types of diseases and many different symptoms, and that these symptoms clustered within families, suggesting that nuclear DNA plays a role in modifying how the mtDNA mutation shows up.”
 research, Dr. Moraes and his lab used one of the base editors to rescue mitochondrial function in a mouse model. “We found a way to base edit a gene with a pathogenic mutation so that it became stable, improving the function of the mitochondrial energy production in the mouse model,” he says.
research, Dr. Moraes and his lab used one of the base editors to rescue mitochondrial function in a mouse model. “We found a way to base edit a gene with a pathogenic mutation so that it became stable, improving the function of the mitochondrial energy production in the mouse model,” he says. a thick skin and to be tenacious. “We have to keep pushing,” he says. “There are lots of failures in this field, but a failure isn’t a total failure if you understand why the experiment didn’t work. It always teaches you something.”
a thick skin and to be tenacious. “We have to keep pushing,” he says. “There are lots of failures in this field, but a failure isn’t a total failure if you understand why the experiment didn’t work. It always teaches you something.”